

Wir haben weitere DNA-Proben von Äschen gesammelt und diese einer neuerliche „Bewertung der natürlichen und gestörten Populationsstruktur der Äsche (Thymallus thymallus) im Salzkammergut“ in Auftrag gegeben um mit einer kombinierte phylogeographische (mitochondriale DNA) und populationsgenetische (Mikrosatelliten) Daten zur Struktur unserer Äschen-Populationen zu erhalten. Die Studie stellte fest , dass es in Populationen südlich der Alpen zu einer umfassenden Genvermischung (Introgression) durch den Menschen kam, während nördlich der Alpen weniger Störungen auftraten.

Hintergrund
Im Jahr 2020/2021 haben wir insgesamt 28 Proben von Äschen aus dem oberen Salzkammergut, darunter war auch Material aus einer Fischzuchtanlagen dabei und haben diese genetisch untersuchen lassen und diese wurden mit verschiedenen Referenzproben aus dem Archiv der Uni-Graz verglichen. Die Ergebnisse dieser Untersuchung aus dem Jahr 2020/2021 haben gezeigt, dass mindestens 10 der 28 Proben, offenbar Abstammungslinien repräsentieren, die eindeutig nicht aus der Region stammen.

Folgestudie 2024/2025
In dieser Folgestudie 2024/2025 werden jetzt 49 neue Proben untersucht, die an verschiedenen Stellen des Traun-Flusssystems im oberen Salzkammergut gesammelt wurden. Diese werden wieder mit Referenzlinien und den Proben aus der vorherigen Studie von 2020/2021 verglichen.
Methodik
Genau wie bei unserer Analyse von 2020/2021, wurden auch mit der neuen Proben-Sammlung 2024 zwei genetische Methoden bei den Proben angewendet.
mtDNA-Analyse
Als Erstes, wurden die sogenannte Kontrollregion der mtDNA-Moleküle sequenziert und in einem zweiten Schritt, die 11 Mikrosatelliten-Loci amplifiziert und auf Allel-Variabilität untersucht. Die mtDNA-Sequenzen helfen dabei, Haplotypen den Hauptlinien zuzuordnen, die schon in mehreren Publikationen Instituts der UNI-Graz aus ganz Europa beschrieben wurden. Dieser Ansatz funktioniert zwar sehr gut, um fremde Abstammungslinien zu erkennen, aber er kann weder die Herkunft einzelner Fische belegen, noch einzelne Fische identifizieren, die gekreuzt wurden. Das liegt daran, dass mtDNA nur über die mütterliche Linie vererbt wird und dass ein Haplotyp, sobald er in eine Population gelangt ist, viele Generationen lang bestehen bleiben kann, auch wenn kein weiteres Fremdmaterial hinzukommt.
Mikrosatelliten-Analyse
Deshalb wird die mtDNA-Analyse mit einer Analyse mittels Mikrosatelliten verbunden, welche eine höhere Auflösung aufweist. Mikrosatelliten werden von beiden Elternteilen vererbt und haben eine höhere Mutationsrate als mtDNA. Dadurch kann man zwischen nah verwandten Populationen bzw. Individuen unterscheiden. Sie liefern Informationen darüber, wie eng die Individuen miteinander verwandt sind und ob ein einzelner Fisch eine Mischung aus mehreren Stämmen ist oder nicht.
mtDNA-Stammbaum
Die Kontroll-Region der mtDNA von 43 Proben wurde erfolgreich sequenziert und wie auch in der früheren Studie, ein Stammbaum mit Referenzproben aus ganz Europa erstellt.
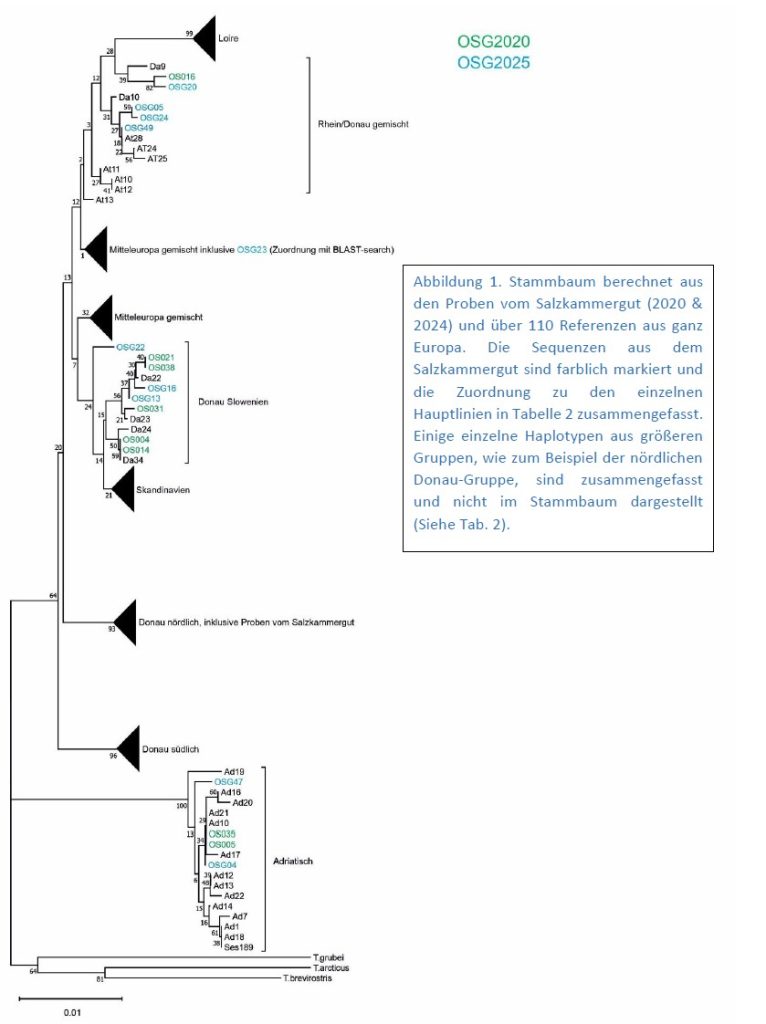
77% einheimischen Linie = Salzkammergut Äsche
Die mtDNA von 33 (77 %) der sequenzierten Individuen, konnte der für die nördliche Alpenregion typischen einheimischen Linie zugeordnet werden. Fünf weitere mtDNA-Sequenzen wurden der sogenannten Rhein/Donau- oder der Mitteleuropa gemischt-Linie zugeordnet, die in der Region auch ziemlich häufig vorkommt.
Die restlichen sechs mtDNA-Sequenzen wurden entweder einer Donau-Slowenien-Linie oder der Adria-Linie zugeordnet und stammen aus Fischbesatzmaßnahmen aus der Aktion „Rettet die Äsche“, die über viele Jahre durchgeführt wurde und 2019 beendet wurde.

Mikrosatelliten Loci Traun Äschen
Mikrosatelliten-Loci sind bestimmte Orte im Genom, die repetitive DNA-Sequenzen wie kurze Tandem-Wiederholungen (STRs) aufweisen, deren Anzahl zwischen Individuen variiert.
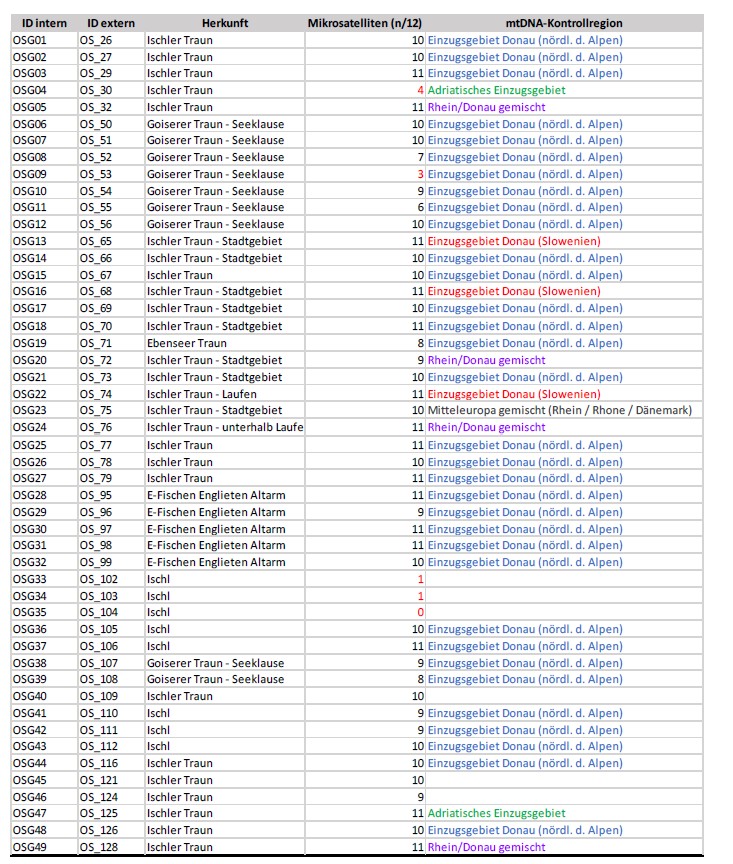
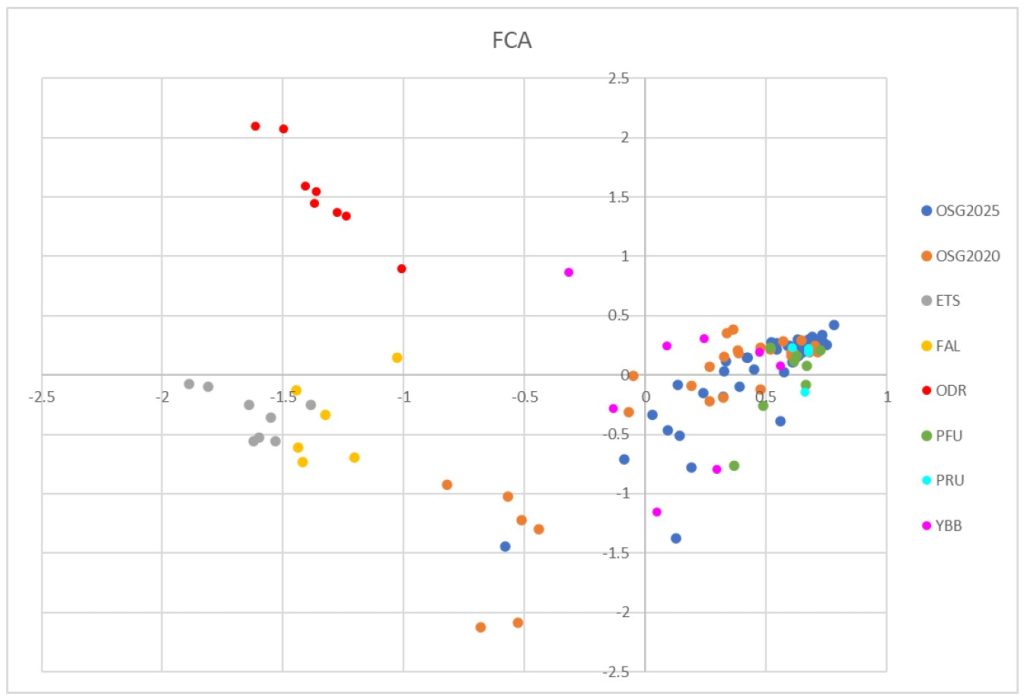
Die Anzahl der erfolgreich amplifizierten Mikrosatelliten-Loci war bei den einzelnen Proben unterschiedlich (siehe Tabelle 2). Die Proben, die nicht in die Analyse einbezogen wurden, sind rot markiert (N = 5, Tabelle 2). Die genetische Beziehung zwischen den einzelnen Proben wird mit Hilfe einer faktoriellen Korrespondenzanalyse dargestellt. Die zu untersuchenden Individuen wurden zusammen mit den sechs Referenzpopulationen, sowie den Proben von 2020 (Tabelle 1) untersucht und die Ergebnisse in Abbildung 2 dargestellt.
Die Grafik zeigt nur die ersten beiden Hauptachsen der Analyse und macht so die größten Unterschiede zwischen den Populationen deutlich, aber nicht unbedingt alle kleinen Unterschiede (dafür könnte man noch weitere Achsen nehmen). Achse 1, trennt nordalpine Individuen, welche auf der rechten Seite der Abbildung zu sehen sind, von allen anderen. Achse 2, trennt wiederum die Linie der Donau, südlich der Alpen (rot, ODR), von den anderen Linien.
Die meisten der Proben aus dem Salzkammergut (2024) liegen in der Gruppe der nördlichen Donaulinie (rechts Mitte). Ein Individuum, fällt allerdings nicht mit diesen und eher mit einer bestimmten Gruppe zusammen, was in der unteren Mitte des Diagramms zu sehen ist. Im Vergleich traf, dies 2020 auf sechs Proben zu.
Auswertung mit Structure
Das Programm „Structure“ ist ein Softwarepaket zur Analyse von Populationsstrukturen anhand von Multilocus-Genotypdaten. Es ermöglicht die Erkennung unterschiedlicher Populationen, die Zuordnung von Individuen zu Populationen, die Untersuchung von Hybridzonen, die Identifizierung von Migranten und gemischten Individuen sowie die Schätzung von Population Allel-Frequenzen in Situationen, in denen viele Individuen Migranten oder gemischte Individuen sind.
Um ein genaueres Verständnis der genetischen Zusammensetzung einzelner Proben zu bekommen, haben wir die Daten noch einer weiteren Analyse, mit dem Programm STRUCTURE unterzogen. Diese Analyse liefert nicht nur eine höhere Auflösung als die ersten beiden Achsen der FCA, sondern gibt auch Aufschluss über den Grad der Vermischung innerhalb einzelner Proben. Diese Analyse wurde in der vorhergehenden Studie 2020/2021 nicht durchgeführt.
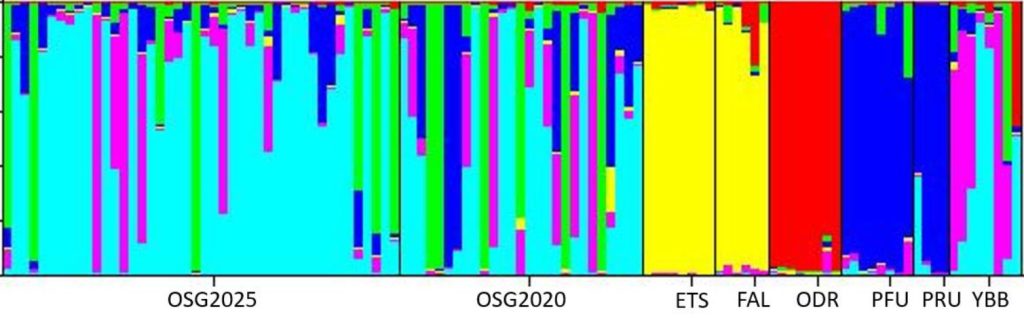
Zum Beispiel gibt’s an der Stelle „Ischler-Traun-Stadt“ hauptsächlich die Population in rosa, während die Population in hellgrün eher an der Stelle „Ischler-Traun“ zu finden ist. Die hellblaue Populationsgruppe kommt im gesamten System vor und könnte daher (spekulativ) eher die einheimische Population repräsentieren.
Diese Analyse zeigt sechs genetische Gruppen, welche unterschiedlich farblich markiert sind.
- Die hellblaue Populationsgruppe kommt im gesamten System vor und könnte daher (spekulativ) die einheimische Population repräsentieren. = Salzkammergut-Äsche
- Die Gelben (ETS & FAL) sind Fische aus dem adriatischen Einzugsgebiet.
- Die Roten (ODR) sind die einheimische südalpine Linie in Österreich.
- Dunkelblauen (PFU & PRU) sind eine Linie aus dem Inn in Tirol.
Bei der Interpretation dieser Gruppen sollte man vorsichtig sein, da sich nicht alle gleich stark voneinander unterscheiden. Analysen mit niedrigeren K-Werten zeigen deutlich, dass die größte Abweichung zwischen ETS & FAL (adriatisch) und allen anderen Proben besteht, wobei sich auch ODR (Südalpen) stärker von den übrigen Populationsgruppen unterscheidet, als die übrigen Populationen voneinander. Die anderen Farben sind eine Mischung aus Populationen von unbekannten Quellen welche sich aber höchstwahrscheinlich zum nördlichen Alpenraum zuweisen lassen. Klar, das Traun-System ist nicht genetisch einheitlich, und die heutige Population wurde wahrscheinlich in der Vergangenheit von mehreren Populationen beeinflusst. Es ist nicht möglich, diese Geschichte zu rekonstruieren, wenn man kein Material von allen möglichen Quellpopulationen hat, die für Zucht- und Besatzprogramme genutzt wurden.

Diskussion
Die Äschen Proben aus dem Einzugsgebiet der Traun von 2024 zeigen ganz klar, ähnlich wie die 2021 analysierten Proben, dass die Population in diesem System nicht einheitlich ist und wahrscheinlich durch Besatzmaßnahmen in der Vergangenheit mit verschiedenen nicht heimischen Populationen vermischt wurde.
- Wenn man aber die möglichen exotischen Abstammungslinien bedenkt, ist die Situation mit den aktuellen Proben im Vergleich zu 2021 besser geworden.
- Mindestens 80 % der Proben von 2024 zeigen einen mtDNA-Haplotyp, der in der Region heimisch ist.
Dabei muss man sich darüber im Klaren sein, dass ausländische mtDNA, sobald sie in den Genpool gelangt ist, wahrscheinlich dortbleibt und noch über viele Generationen nachweisbar sein wird.
Aus der Mikrosatellitenanalyse, vor allem mit „STRUCTURE“, geht klar hervor, dass bestimmte Genotypen aus verschiedenen Populationen an manchen Probenahmestellen häufiger vorkommen als an anderen.

Anhang 1 – Genetische Marker
- mtDNA
Die Analyse eines Abschnitts (Kontroll-Region) der mitochondriellen DNA erlaubt die Differenzierung zwischen verschiedenen Linien durch Zuordnung zu bekannten Haplotypen. Diese Methodik ermöglicht eine eindeutige Zuordnung des mtDNA-Moleküls zu einer dieser Linien, ist aber durch die mütterliche Vererbung der mtDNA limitiert. In Populationen, in welchen Introgression (Vermischung) stattgefunden hat, ist dieser Marker demnach nur ein Anhaltspunkt. - Mikrosatelliten
Es wurden zwölf bei Thymallus thymallus untersucht, die im Rahmen von Untersuchungen an europäischen Äschen (Weiss et al 2013) optimiert wurden. Sie besitzen eine sehr hohe Mutationsrate und werden bi-parental vererbt, weshalb sie den Genfluss der weiblichen als auch der männlichen Elterntiere reflektieren. So wird eine Bestimmung der Verwandtschaftsverhältnisse, sowohl von Einzelindividuen zueinander, als auch von der gesamten Population zu anderen auto- oder allochthonen Populationen aus unserem Datensatz ermöglicht. Aus unserer hausinternen Referenzdatenbank haben wir Referenzpopulationen ausgewählt, die für bestimmte Linien als Vergleichsbasis dienen (Tabelle 1). - Auswahl der Methodik
Sequenzen der mtDNA (Haplotypen) haben den Nachteil, einer rein weiblichen Vererbungslinie, bringen aber den großen Vorteil mit, dass sie über eine lange Zeitspanne (1000en von Jahren) in einer Population stabil bleiben. Dadurch können sie als genetischer Marker für vergangene Durchmischung von natürlichen und fremden Linien dienen. Auf der anderen Seite zeigen Mikrosatelliten mit ihrer bi-parentalen Vererbung ein kompletteres Bild der genetischen Verwandtschaft von Individuen, besonders innerhalb von Populationen. Allerdings können die Anzeichen einer Durchmischung nach einigen Generationen verloren gehen und Verwandtschaften mit sehr weit entfernten Linien (z.B. von Skandinavien) dadurch übersehen werden. Diese beiden Merkmale werden unabhängig voneinander vererbt, was dazu führt, dass die Ergebnisse der beiden Marker für ein Individuum nicht zwangsläufig übereinstimmen müssen. Kombiniert man diese beiden genetischen Marker, bietet sich in Kombination ein bestmögliches Bild von sowohl historischem, als auch gegenwärtigem Genfluss zwischen verschiedenen Linien.

Wichtigste Erkenntnisse
- Störung durch Besatz: Durch menschliche Besatzmaßnahmen kam es zu einer erheblichen genetischen Durchmischung der Äschenpopulationen südlich der Alpen.
- Begrenzte Störungen nördlich der Alpen: Populationen nördlich der Alpen zeigten weniger Anzeichen einer langfristigen Genvermischung aus allochthonen (nicht heimischen) Beständen.
- Genetische Korrelation: Die Studie ergab eine positive Korrelation zwischen mtDNA-Beimischung und genetischer Vielfalt, was die jüngste Genvermischung und das Fehlen starker reproduktiver Barrieren zwischen den Hauptlinien unterstützt.
- Auswirkungen auf den Naturschutz: Die Ergebnisse verdeutlichten eine Abweichung von bestehenden Modellen evolutionär bedeutsamer Einheiten für Thymallus thymallus in Europa.
Ergänzende Informationen zu Genvarianten

Die Allel-Variabilität
Allel-Variabilität beschreibt das Vorkommen unterschiedlicher Allelformen (Genvarianten) an einem bestimmten Genort in einer Population, was zu genetischer Diversität führt. Sie ist die Grundlage für die Vielfalt von Merkmalen bei Organismen und entsteht durch Prozesse wie Mutationen, die neue Allele hervorbringen, und Rekombination, die vorhandene Allele neu mischt.
Allel-Variabilität ist essenziell für die Anpassung von Arten an ihre Umwelt
und die treibende Kraft hinter der natürlichen Selektion.
Was ist ein Allel?
- Ein Allel ist eine spezifische Version eines Gens, das sich an einer bestimmten Stelle (Locus) auf einem Chromosom befindet.
- Verschiedene Allele eines Gens können zu unterschiedlichen physischen oder physiologischen Merkmalen führen.
Was ist Variabilität?
- Variabilität ist die Vielfalt von Merkmalen innerhalb einer Population.
- Wenn es um genetische Variabilität geht, sind dies die Unterschiede in der genetischen Information, die durch die verschiedenen Allele verursacht werden.
Wie entsteht Allel-Variabilität?
- Mutationen: Zufällige Veränderungen in der DNA-Sequenz sind die Quelle neuer Allele, die dann die genetische Vielfalt erhöhen.
- Rekombination: Während der Meiose (sexuelle Fortpflanzung) werden vorhandene Allele neu gemischt, was zu neuen Kombinationen und somit zu genetischer Vielfalt führt.
Bedeutung der Allel-Variabilität
- Anpassung: Eine große allelische Variabilität in einer Population ermöglicht es der Art, sich besser an veränderte Umweltbedingungen anzupassen.
- Natürliche Selektion: Individuen mit vorteilhaften Allelen können besser überleben und sich vermehren, was die genetische Struktur der Population im Laufe der Zeit verändert.
- Messung der genetischen Diversität: Durch die Untersuchung der Allelfrequenzen (der Prozentsatz eines bestimmten Allels in einer Population) können Wissenschaftler die genetische Vielfalt messen.

Quellenverweis
Anhang 2 – Referenzen
Assessing natural and disturbed population structure in European grayling Thymallus thymallus: melding phylogeographic, population genetic and jurisdictional perspectives for conservation planning. Journal of Fish Biology 82, 505-521 von Weiss SJ, Kopun T, Sušnik Bajec S. (2013).
Genetische Untersuchungen an Proben der Europäischen Äsche (Thymallus thymallus) aus dem oberen Salzkammergut und ausgewählten Zuchten. Endbericht im Auftrag des Fischereirevier Oberes Salzkammergut. 7 Seiten von Weiss S, Frühlich D. (2021).
Weitere Informationen
Ein passendes Zitat zum Thema Fischbesatz:
„An allem Unfug der passiert, sind nicht etwa nur die schuld, die ihn tun, sondern auch die, die ihn nicht verhindern.“
Zitat von: Erich Kästner 